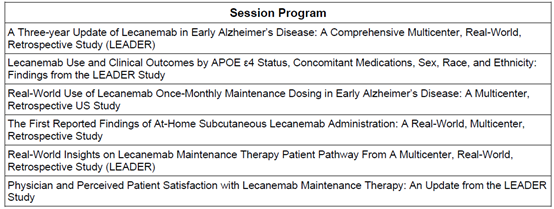
美国FDA批准乐意保®(仑卡奈单抗)皮下自动注射剂型用于早期阿尔茨海默病初始治疗
乐意保®皮下自动注射剂型是全球首个获批可支持患者居家完成初始给药与长期维持治疗抗淀粉样蛋白治疗药物。
乐意保®皮下自动注射剂型计划于 2026 年 8 月下旬在美国上市。
2026 年 7 月 14 日,东京与马萨诸塞州剑桥讯 —— 卫材株式会社与渤健公司(纳斯达克股票代码:BIIB)联合宣布,美国食品药品监督管理局(FDA)已批准仑卡奈单抗(lecanemab-irmb)每周一次皮下注射制剂(中文商品名:乐意保®,美国商品名:LEQEMBI IQLIK®)的补充生物制品许可申请(sBLA),用于早期阿尔茨海默病的初始给药治疗。
乐意保®皮下自动注射剂型采用自动注射器给药,为患者治疗初期提供了相较于静脉输注的新的给药选择。获批初始给药方案为 500 毫克/周,分两次,每次 250 毫克注射,单次注射时长约 15 秒。
在从仑卡奈单抗初始治疗到维持治疗的整个疗程中,患者可根据治疗需要采用静脉输注或皮下注射两种给药方式,也可在两种给药方式间灵活切换,提高用药的便捷性与灵活性。
仑卡奈单抗在美国的获批适用人群为:因阿尔茨海默病引发轻度认知障碍或轻度痴呆的成年患者(以上病症统称为早期阿尔茨海默病)。由阿尔茨海默病引起的轻度认知障碍是阿尔茨海默病最早出现症状的阶段,可能表现为细微的症状,如记忆力减退、思维混乱、言语卡顿等。
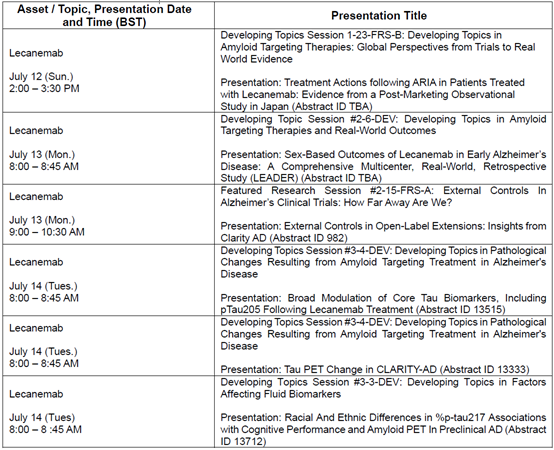
FDA批准皮下注射初始给药方案的临床数据支撑
FDA 批准乐意保®作为初始给药方案,依托于一套完整临床数据体系:多项研究评估了在不同给药剂量方案下仑卡奈单抗皮下给药的疗效与安全性。本次获批核心依据来自Ⅲ期Clarity AD临床试验18个月核心研究后的长期扩展子研究,针对早期阿尔茨海默病患者的试验结果如下:
- 每周一次皮下注射的药物暴露量与静脉输注方案相当,支持获得相似的临床疗效及生物标志物获益;
- 皮下自动注射剂型给药后,ARIA-E等相关不良事件的发生率预计与静脉给药相似。相较于安慰剂,LEQEMBI未提高孤立性ARIA-H(指未合并ARIA-E的患者中发生的ARIA-H)的发生率;
- 使用 LEQEMBI 皮下注射制剂时观察到注射相关反应,其中绝大多数为局部反应,而全身性反应的发生频率相对较低。
阿尔茨海默病药物发现基金会(ADDF)联合创始人、荣誉首席科学Howard Fillit博士表示:“乐意保®皮下自动注射剂型获批用于初始给药治疗,标志着阿尔茨海默病治疗进入新的阶段。患者及其照护者首次能够自主选择抗淀粉样蛋白疗法的给药方式。随着治疗手段的不断丰富,给药技术的创新将发挥关键作用,有助于提升药物可及性、推动潜在联合治疗方案的研究,并助力阿尔茨海默病精准医疗模式的发展。”
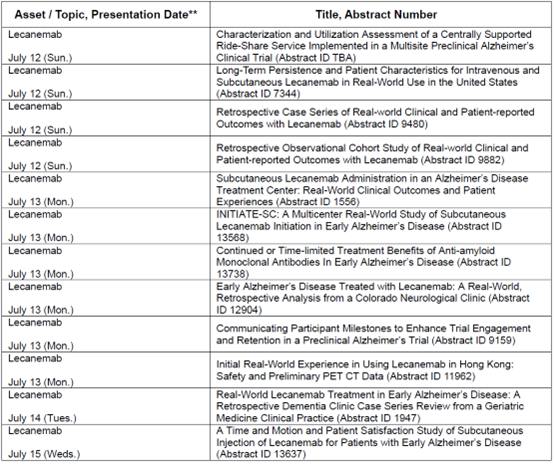
全面提升阿尔茨海默病全程诊疗的治疗灵活性
乐意保®皮下自动注射剂型初始给药方案的获批,为患者及照护者提供了目前获美国FDA批准的可贯穿阿尔茨海默病全程治疗的居家给药方案,能够适配不同医疗场景,有望支持诊疗流程优化,并改善药物可及性。皮下注射给药可实现多项潜在获益:
- 减少患者与照护者往返医院就诊的负担
- 降低静脉输注需求,节约配套医疗资源
- 缩短治疗准备、给药时长与护士监护需求
- 给偏好或必须接受静脉输注治疗的患者保留充足的临床输注医疗资源。
一项自动注射器使用认可度研究数据显示:94%的早期阿尔茨海默病患者及其照护者认为乐意保®皮下自动注射剂型给药装置操作简便,且对居家用药的满意度和使用信心均处于较高水平。*
*基于对50名早期阿尔茨海默病(AD)患者和50名早期AD患者的照护者进行的面对面访谈。
参与者有机会实际操作一款训练用自动注射器(不含针头和药液)及注射垫,随后被要求在计算机上完成问卷调查,反馈其使用体验,其中包含“您觉得使用该自动注射器的难度如何?”等问题。
媒体联络 卫材株式会社 公关部 电话:+81 (0) 3-3817-5120
卫材欧洲有限公司 欧洲、中东、非洲通讯部 电话:+44 (0) 7760 619251 邮箱:Emea-comms@eisai.net
卫材美国公司 Libby Holman 电话:+1-201-753-1945 邮箱:Libby_Holman@Eisai.com
渤健公司 Madeleine Shin 电话:+1-781-464-3260 邮箱:public.affairs@biogen.com
投资者联络 卫材株式会社 投资者关系部 电话:+81 (0) 3-3817-5122 渤健公司 Tim Power 电话:+1-781-464-2442 邮箱:IR@biogen.com
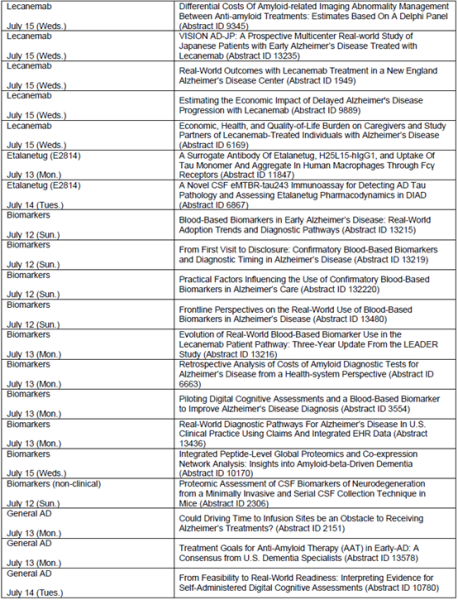
参考资料
关于卫材与渤健在阿尔茨海默病(AD)治疗领域的合作
卫材与渤健自2014年起,便在阿尔茨海默病治疗药物的联合开发及商业化方面展开合作。卫材负责全球范围内仑卡奈单抗(lecanemab)的研发主导及注册申报工作,双方共同进行该产品的商业化及推广活动,卫材拥有最终决策权。

副校(院)长贾海宁发表致辞")